血小板增多可以活多久?

上周有个女孩咨询,血小板发现增高半年,为^9/L,患者比较焦虑,这个可以理解,她没有任何症状,是体检发现的,一开始^9/L,后来慢慢升高。那么这个病是什么情况,如何诊断,预后如何呢?
关于诊断
原发性血小板减少症(ET)患者血小板计数明显增高,多数在^9/L以上。血涂片可见成堆的血小板。骨髓巨核细胞增多,体积增大并呈丛集分布。与JAK2 V617F突变阳性患者比较,CALR基因突变患者的血小板水平较高,而血红蛋白水平与白细胞计数较低一。MPL基因突变的ET患者的血小板计数也较高、血红蛋白水平较低、骨髓增生程度较低。JAK2 V617F基因突变检测已成为诊断ET的常规实验室项目,CALR与MPL基因突变检测也正在逐渐推广。当然JAK2 V617F基因的特异性不高血小板增多,敏感性较高,骨髓增殖性疾病还有其他正常人也有检出,需要结合血小板的情况。所以本病的诊断不难。
目前国内外普遍采用2008年WHO修订的诊断标准,主要内容为:①血小板计数持续>^9/L;②骨髓检查示巨核系增生,以成熟的大巨核细胞数量增加为主,无明显粒系或红系增生;③不符合慢性髓性白血病(CML)、真性红细胞增多症(PV)、骨髓纤维化(MF)、骨髓增生异常综合征(MDS)或其他骨髓增殖性疾病的WHO诊断标准;④有JAK2 V617F基因或其他克隆标志的表达或无反应性血小板增多的证据。诊断要求符合上述所有4项标准。WHO修订的MPN诊断标准包括了JAK2基因第12号外显子与MPL基因突变。目前已有很多中心将CALR基因突变列为ET与MF诊断新的分子学指标。有理由相信,在未来WHO新的修订的诊断标准过程中也会将CALR补充列入。
关于鉴别
血小板计数在很多病理状态下都可能明显增高,有时与ET难以鉴别。CML呈特异的Ph染色体与BCR-ABL融合基因阳性。ET患者很少有血红蛋白水平增高,男性患者HGB>165 g/L、女性患者HGB>160 g/L时可基本排除ET,而应诊断为PV。ET仅有轻度骨髓网状纤维组织增生,在有明显纤维化时应诊断为MF。此外,反应性血小板增多可见于缺铁性贫血与失血、慢性感染、组织损伤、慢性炎症、肿瘤、溶血与脾切除后等。ET在儿童相当少见。在血小板增多的儿童中,仅1/4确定有相关的基因突变,因此对儿童要首先考虑反应性血小板增多。
预后
这个是大家最关心的。 ET虽属造血系统恶性克隆性疾病血小板增多,但只要控制外周血血小板水平并预防血栓和出血并发症,患者的寿命可基本不受影响。所以,基本不受影响的意思不言自明,所以其实大可不必太担心,至于血栓和出血,其实正常人也很多见,所以,我经常把这个病根高血压和糖尿病相比较,来让患者了解。有CALR突变的ET患者平均生存期可达20.2年,但如合并有ASXLl突变则预后不良;无、CALR、MLP基因突变的“三阴性”ET患者生存期反而较短。
治疗
这个其实大家也比较关心,我们目前的观点,如果发现血小板升高,在400-^9/L,可以仅服用阿司匹林,或者联合活血化瘀中药;血小板在500-^9/L,可以服用阿司匹林联合活血化瘀中药;如果血小板在600-^9/L,除了以上治疗,还需要联合其他中成药,比如脑血康、大黄蛰虫丸、心脑欣丸等;如果血小板在^9/L,应该先用羟基脲或者干扰素让血小板降低到相对稳定水平,再按照上述方案治疗。这样做的直接目的就是提高生活质量。



随便看看
怎样加入汽车之家比亚迪唐论坛 宝马汽车论坛怎么进入
2024-03-25
刘鸣炜华人置业盈利预警,身价135亿美元!
2024-03-26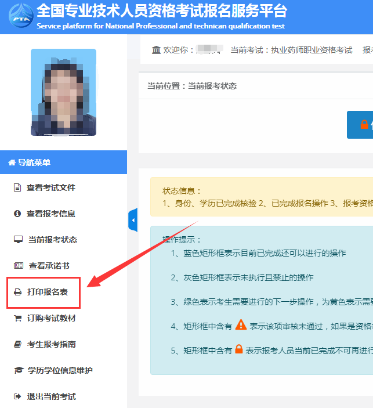
2021年执业药师考试报名表的模板+填写规范!
2024-03-25
康迪k10b与k11的区别 康迪k11能加电池吗
2024-03-25来也来去也去不是滚滚红尘还有甚么歌 有一首歌中有一句:来呀来去啊去这是什么歌
2024-03-22消逝的光芒2克兰怎么样了 什么是克兰银兰
2024-03-21
维生素AD和维生素D到底有啥区别?看完你就知道了
2024-03-27仙剑奇侠传1几个结局 仙剑奇侠的结局是什么
2024-03-24